Gizli Maküler Distrofi / Occult Macular Dystrophy
Gizli Maküler Distrofi
Occult Macular Dystrophy
Işıl Sayman Muslubaş, Serra Arf, Mümin Hocaoğlu, Hakan Özdemir, Murat Karaçorlu
İstanbul Retina Enstitüsü, İstanbul, Türkiye
İstanbul Retina Enstitüsü, İstanbul, Türkiye
GİRİŞ
Gizli maküler distro (GMD), her iki gözde ilerleyici görme kaybıileseyredennormalfundusgörünümüilekarakterizekalıtsal makülar bir distro dir.1 Hastalarda fundus resein anjiogra (FFA) incelemesi ve tüm alan elektroretinogram (ERG) tetkiki normal olmakla beraber merkezi retina tetkiki olan multifokal ERG (mfERG) sonuçları belirgin şekilde düşüktür.1,2 Spektral domain optik koherens tomogra (SD-OKT) incelemesinde foveal kalınlıkta azalma ile birlikte fotoreseptör iç ve dış segment birleşiminde (IS/OS) bozulma gözlenir. Pek çok hastada SD-OKT ile tespit edilen fotoreseptör tabakasındaki bozulma, görme keskinliği ve hastalığın progresyonu ile uyumludur.3 Mikroperimetri (MP) maküla hastalıklarında retina hassasiyetini belirlemek için kullanılan bir görme alanı tekniğidir.4 Merkezi retinayı ilgilendiren hastalıklarda skotom ve görme kayıplarına bağlı ksasyon değişikliklerini belirlemede gelişmiş bir yöntemdir ve GMD’li hastalarda bu amaçla kullanılabilir. 5,6,7 Fundus infrared re ektans (İR) görüntülüme tekniğinde GMD’li hastalarda santral hipore ktans görülür ve hastalığın ilerlemesiyle ktans görünüm daha belirgin hale gelir. 8 Fundus oto sans (FOF) görüntüleme tekniğinde ise GMD’li çoğu hastada belirgin bir anormallik gözlenmezken az sayıda hastada f santral hiper sans görülebilir. 8,9
Bu olgu sunumunda GMD tanısı koyduğumuz bir hastanın klinik özelliklerini ve tanı yöntemlerini sunmayı amaçladık.
Olgu Sunumu
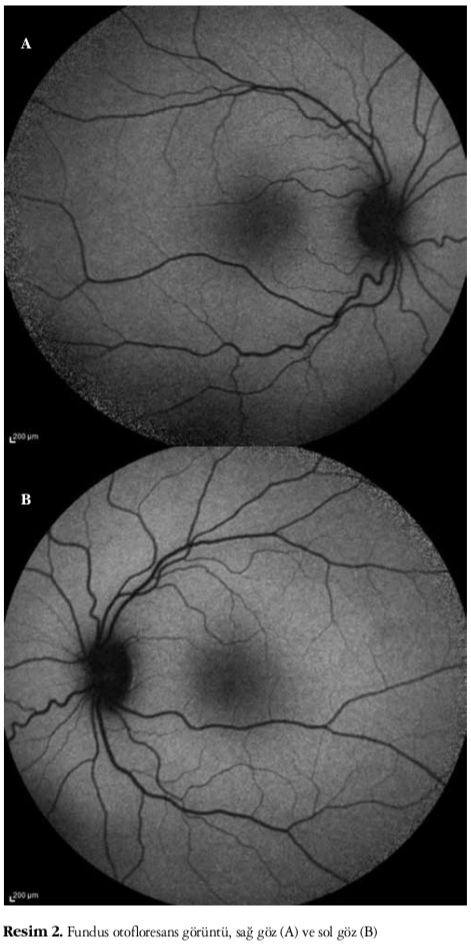
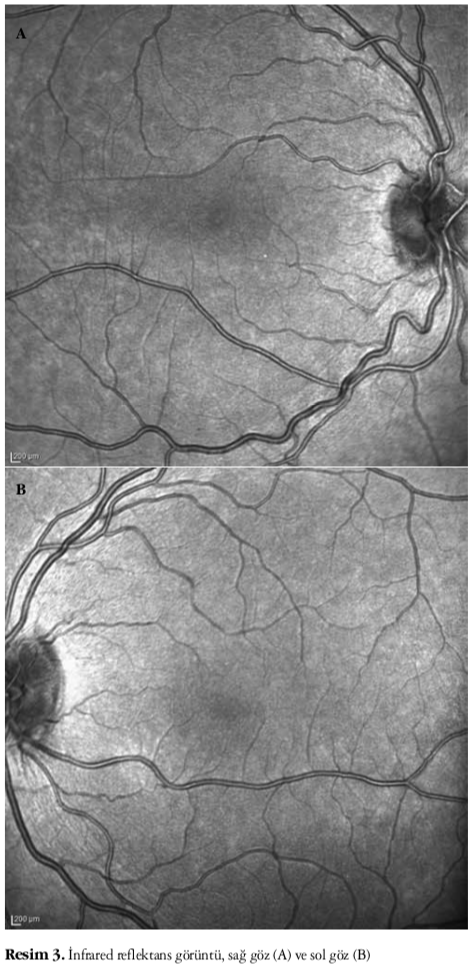
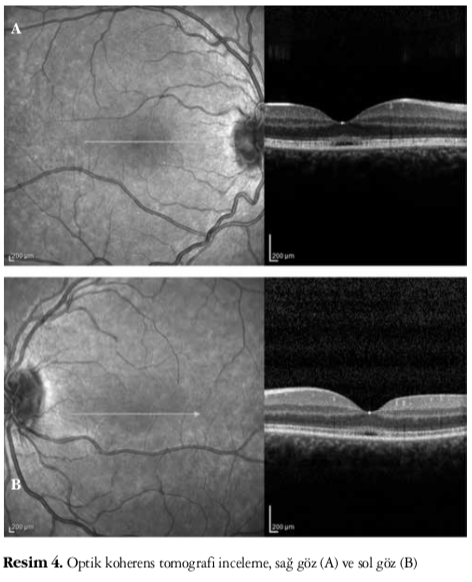
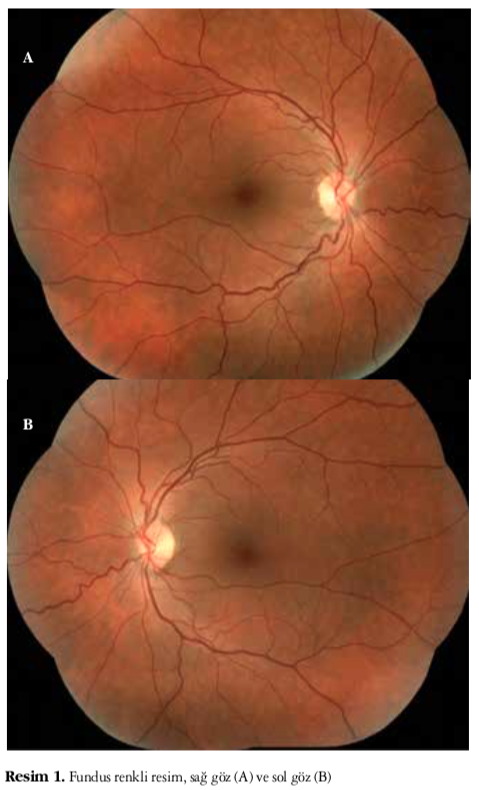 Altı yıldır her iki gözde ilerleyici görme kaybı olan 20 yaşındaki kadın hasta kliniğimize refere edildi. Hastanın bilinen bir sistemik hastalığı, travma öyküsü, aile hikayesi, ilaç ya da sigara kullanım öyküsü ve anne-baba akraba evliliği yoktu. Yapılan muayenesinde görme keskinliği her iki gözde 3-4/10 düzeyinde, göz içi basınçları sağda 11 mmHg, solda 13 mmHg olarak bulundu. Ön segment muayenesinde bir patoloji tespit edilmedi. Yapılan fundus muayenesinde retina damarlarında hafif bir kıvrımlanma artışı dışında bir patoloji tespit edilmedi (Resim 1A ve 1B). Ishihara renk görme testinde yakından bakarak tüm plakaları okudu. FOF incelemesi her iki gözde normaldi (Resim 2A ve 2B). Fundus İR görüntüleme tekniği ile her iki gözde merkezi hafif hiporeflektans görüldü (Resim 3A ve 3B). FFA incelemesi normaldi. OKT incelemesi kalınlık profili analizinde foveal kalınlık sağ gözde 155 μm ve sol gözde 188 μm olarak tespit edildi. Fotoreseptör IS/OS birleşiminde bozulma gözlendi. IS/OS bandındaki bozulmanın yatay eksendeki yayılımı incelendiğinde sağ gözde 696 μm ve sol gözde 348 μm olarak
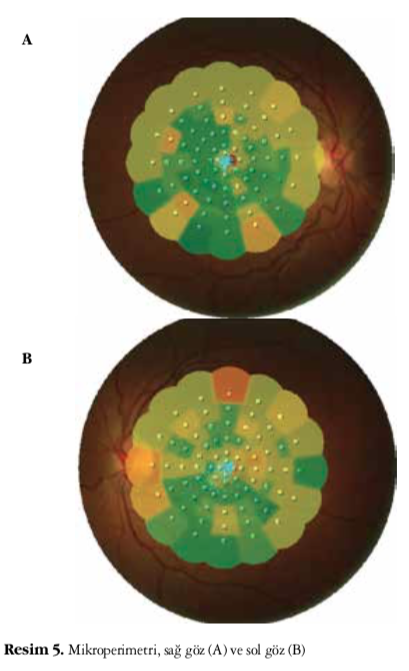
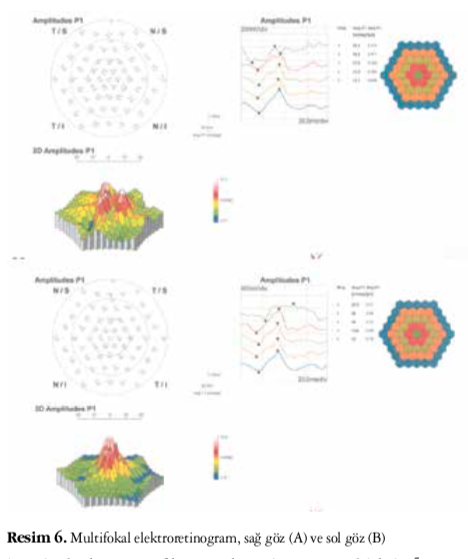
ölçüldü (Resim 4A ve 4B). MP tetkikinde yine sağ gözde daha belirgin olmakla birlikte her iki gözde rölatif stabil olmayan fiksasyon ve OKT ile uyumlu olan bölgelerde absolü skotom tespit edildi. Retinal duyarlılık, maküla 20 derecelik alanda sağ gözde 13,9 dB ve sol gözde 13,5 dB olarak ölçüldü (Resim 5A and 5B). Tüm alan ERG incelemesi normal bulunurken mfERG incelemesinde sağ gözde daha belirgin olmakla birlikte her iki gözde santral cevabın azaldığı gözlendi (Resim 6A and 6B).
Altı yıldır her iki gözde ilerleyici görme kaybı olan 20 yaşındaki kadın hasta kliniğimize refere edildi. Hastanın bilinen bir sistemik hastalığı, travma öyküsü, aile hikayesi, ilaç ya da sigara kullanım öyküsü ve anne-baba akraba evliliği yoktu. Yapılan muayenesinde görme keskinliği her iki gözde 3-4/10 düzeyinde, göz içi basınçları sağda 11 mmHg, solda 13 mmHg olarak bulundu. Ön segment muayenesinde bir patoloji tespit edilmedi. Yapılan fundus muayenesinde retina damarlarında hafif bir kıvrımlanma artışı dışında bir patoloji tespit edilmedi (Resim 1A ve 1B). Ishihara renk görme testinde yakından bakarak tüm plakaları okudu. FOF incelemesi her iki gözde normaldi (Resim 2A ve 2B). Fundus İR görüntüleme tekniği ile her iki gözde merkezi hafif hiporeflektans görüldü (Resim 3A ve 3B). FFA incelemesi normaldi. OKT incelemesi kalınlık profili analizinde foveal kalınlık sağ gözde 155 μm ve sol gözde 188 μm olarak tespit edildi. Fotoreseptör IS/OS birleşiminde bozulma gözlendi. IS/OS bandındaki bozulmanın yatay eksendeki yayılımı incelendiğinde sağ gözde 696 μm ve sol gözde 348 μm olarak
ölçüldü (Resim 4A ve 4B). MP tetkikinde yine sağ gözde daha belirgin olmakla birlikte her iki gözde rölatif stabil olmayan fiksasyon ve OKT ile uyumlu olan bölgelerde absolü skotom tespit edildi. Retinal duyarlılık, maküla 20 derecelik alanda sağ gözde 13,9 dB ve sol gözde 13,5 dB olarak ölçüldü (Resim 5A and 5B). Tüm alan ERG incelemesi normal bulunurken mfERG incelemesinde sağ gözde daha belirgin olmakla birlikte her iki gözde santral cevabın azaldığı gözlendi (Resim 6A and 6B).
Tartışma
 GMD, kalıtımsal bir maküler distrofidir ve maküler bir disfonksiyon olmasına rağmen fundus görünümü normal olduğu için gizli olarak adlandırılmıştır. İlk olarak Miyake ve ark.1 tarafından tanımlanan bu hastalık otozomal dominant geçişli olsa da sporodik olgular da bildirilmiştir.
GMD, kalıtımsal bir maküler distrofidir ve maküler bir disfonksiyon olmasına rağmen fundus görünümü normal olduğu için gizli olarak adlandırılmıştır. İlk olarak Miyake ve ark.1 tarafından tanımlanan bu hastalık otozomal dominant geçişli olsa da sporodik olgular da bildirilmiştir.
Hastalığın başlangıcının 6 ile 81 yaş arasında geniş bir aralıkta olduğu birçok yayında bildirilmiştir.9 Bizim olgumuzda görme kaybı 14 yaşında başlamakla beraber tanısı 20 yaşında konulmuştur.
GMD merkezi retina hastalığı olduğu için hastaların tüm alan ERG tetkiki normal olmakla beraber fokal maküler ERG ve mfERG sonuçları belirgin olarak düşüktür.1,2 Ayrıca merkezi retina duyarlılığını ölçen MP testinde de GMD’li hastalarda skotom ve fiksasyon kayıpları tespit edilebilir.5 Bizim olgumuzda diğer yayınlarda da belirtildiği üzere tüm alan ERG sonuçları normal tespit edilirken mfERG tetkikinde sağ gözde daha belirgin olmak üzere her iki gözde santral cevabın azaldığı gözlendi. MP testinde de yine sağ gözde daha belirgin olmak üzere her iki gözde rölatif stabil olmayan fiksasyon tespit edildi. Maküla 8 derecelik alanda retinal hassasiyetin her iki gözde de azaldığı görüldü.
SD-OKT ile GMD’li hastalarda fotoreseptör tabakasındaki yapısal değişiklikler kolayca tespit edilebilir. Birçok yayında SD-OKT ile görüntülemede hastalarda foveada belirgin fotoreseptör hasarı, fovea kalınlığında azalma ve fotoreseptör IS/ OS birleşimde bozulma bildirilmiştir.3
Fotoreseptör tabakasındaki bozulma derecesi ile görme keskinliği ve hastalığın ilerleyişi arasında da bir korelasyon olduğu gösterilmiştir.3 Olgumuzda da benzer olarak sağ gözde daha belirgin olmak üzere her iki gözde fovea kalınlığında azalma ve fotoreseptör IS/OS birleşiminde bozulma gözlendi. Bir diğer görüntülüme yöntemi olan fundus İR inceleme kolay uygulanabilir olması, GMD’li hastalarda merkezi hiporeflektans göstermesi ve hastalığın progresyonunda bu hiporeflektans görünümün daha belirginleşmesi nedeniyle tanıda yardımcı bir yöntem olarak kullanılabilir.8,9,10,11 GMD’de asıl problem fotoreseptörlerde olduğundan ve de retina pigment epitelinde belirgin bir hasar olmadığından FOF incelemesinde belirgin bir anormallik saptanmaz.9 Olgumuzda da FOF incelemesi normalken, fundus İR görüntüleme tekniğinde merkezi hafif hiporeflektans tespit edilmiştir.
Sonuç
Sonuç olarak, ilerleyici görme kaybı olup fundus görünümü ve FFA tetkiki normal olan, klinik olarak GMD ile uyumlu hastalarda, SD-OKT, invazif olmayan, kolay uygulanabilen, öncelikli ve kliniği daha güvenilir bir araçtır. Fundus İR, mfERG ve MP yardımcı diğer tanı yöntemleridir.
Etik
Hasta Onayı: Helsinki deklarasyon prensiplerine uygunluk ilkesine bağlı kalınarak, mevcut durum ve doğal seyir hakkında bilgi verilerek hasta onamı alınmıştır.
Hakem Değerlendirmesi: Editörler kurulu dışında olan kişiler tarafından değerlendirilmiştir.
Yazarlık Katkıları
Cerrahi ve Medikal Uygulama: Serra Arf, Hakan Özdemir, Murat Karaçorlu, Konsept: Işıl Sayman Muslubaş, Serra Arf, Hakan Özdemir, Murat Karaçorlu, Dizayn: Işıl Sayman Muslubaş, Serra Arf, Murat Karaçorlu, Veri Toplama veya İşleme: Işıl Sayman Muslubaş, Mümin Hocaoğlu, Analiz veya Yorumlama: Işıl Sayman Muslubaş, Serra Arf, Murat Karaçorlu, Literatür Arama: Işıl Sayman Muslubaş, Mümin Hocaoğlu, Yazan: Işıl Sayman Muslubaş, Mümin Hocaoğlu, Serra Arf, Hakan Özdemir, Murat Karaçorlu. Çıkar Çatışması: Yazarlar tarafından çıkar çatışması bildirilmemiştir.
Finansal Destek: Yazarlar tarafından finansal destek almadıkları bildirilmiştir.
Kaynaklar
1.Miyake Y, Ichikawa K, Shiose Y, Kwase Y. Hereditary macular dystrophy without visible fundus abnormality. Am J Ophthalmol. 1989;108:292-299.
2. Piao CH, Kondo M, Tanikawa A, Terasaki H, Miyake Y. Multifocal electroretinogram in occult macular dystophy. Invest Ophthalmol Vis Sci. 2000;41:513-517.
3. Park SJ, Woo SJ, Park KH, Hwang JM, Chung H. Morphologic photoreceptor abnormality in occult macular dystrophy on spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2010;51:3673-3679.
4. Rohrschneider K, Bültmann S, Springer C. Use of perimetry (microperimetry) to quantify macular sensitivity. Prog Retin Eye Res. 2008;27:536-548.
5. Freund PR, Macdonald IM. Microperimetry in a case of occult macular dystrophy. Can J Ophthalmol. 2013;48:101-103.
6. Şentürk F, Karaçorlu SA, Özdemir H, Karaçorlu M, Uysal Ö. Klasik ve gizli koroid neovaskülarizasyonlarında mikroperimetrik değişiklikler. Ret-Vit. 2007;15:277-281.
7. Şentürk F, Özdemir H, Karaçorlu S, Karaçorlu M. Stargardt hastalığında fiksasyon özelliklerinin MP-1 mikroperimetri ile değerlendirilmesi. Turk J Ophthalmol. 2008;38:134-138.
8. Ahn SJ, Ahn J, Park KH, Woo SJ. Multimodal imaging of occult macular dystrophy. JAMA Ophthalmol. 2013;131:880-890.
9. Fujinami K, Tsunoda K, Hanazono G, Shinoda K, Ohde H, Miyake Y. Fundus autofluorescence in autosomal dominant occult macular dystrophy. Arch Ophthalmol. 2011;129:597-602.
10. Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, Usui T, Hatase T, Nakamura M, Ohde H, Itabashi T, Okamoto H, Takada Y, Iwata T. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet. 2010;87:424-429.
11. Ahn SJ, Cho SI, Ahn J, Park SS, Park KH, Whoo SJ. Clinical and genetic characteristics of Korean occult macular dystrophy patients. Invest Ophthalmol Vis Sci. 2013;54:4856-4863.





